MSanalysis - A Push-based System for Molecular Simulation Data Analysis
Molecular Simulation can provide insights into the structure, dynamics, and thermodynamic characteristics of biological structures. Computer simulation is an important method for molecular dynamics research. Many scientific fields generate, and require manipulation of big data. Known as scientific data. The outcome of an MD simulation is a "pseudo trajectory" of positions and velocities of all of the atoms in the simulation system. Due to the large number of atoms/molecules and time instances to simulate, such simulations generate tremendous amounts of data. How to effectively and efficiently reason about such data is a critical problem that directly affects the success of any molecular simulation study.

Traditional MS analysis software (e.g., GROMACS, NAMD, AMBER) are widely used in many scientific domains. However, these existing software fall short in handling simulation outputs that come with increasingly high volume. They follow a pull-based architectural design, where the executed queries mandate the data needed. Such design involves huge amount of redundant and random I/Os. In this project, we design and implement MSanalysis, a push-based system that allows high-throughput data analysis in the process of scientific discovery. Our design improves throughput in two ways: i) it uses a sequential scan-based I/O framework that loads the data into the main memory only once, and then ii) the system pushes the loaded data to a number of pre-programmed queries and more than one selections.
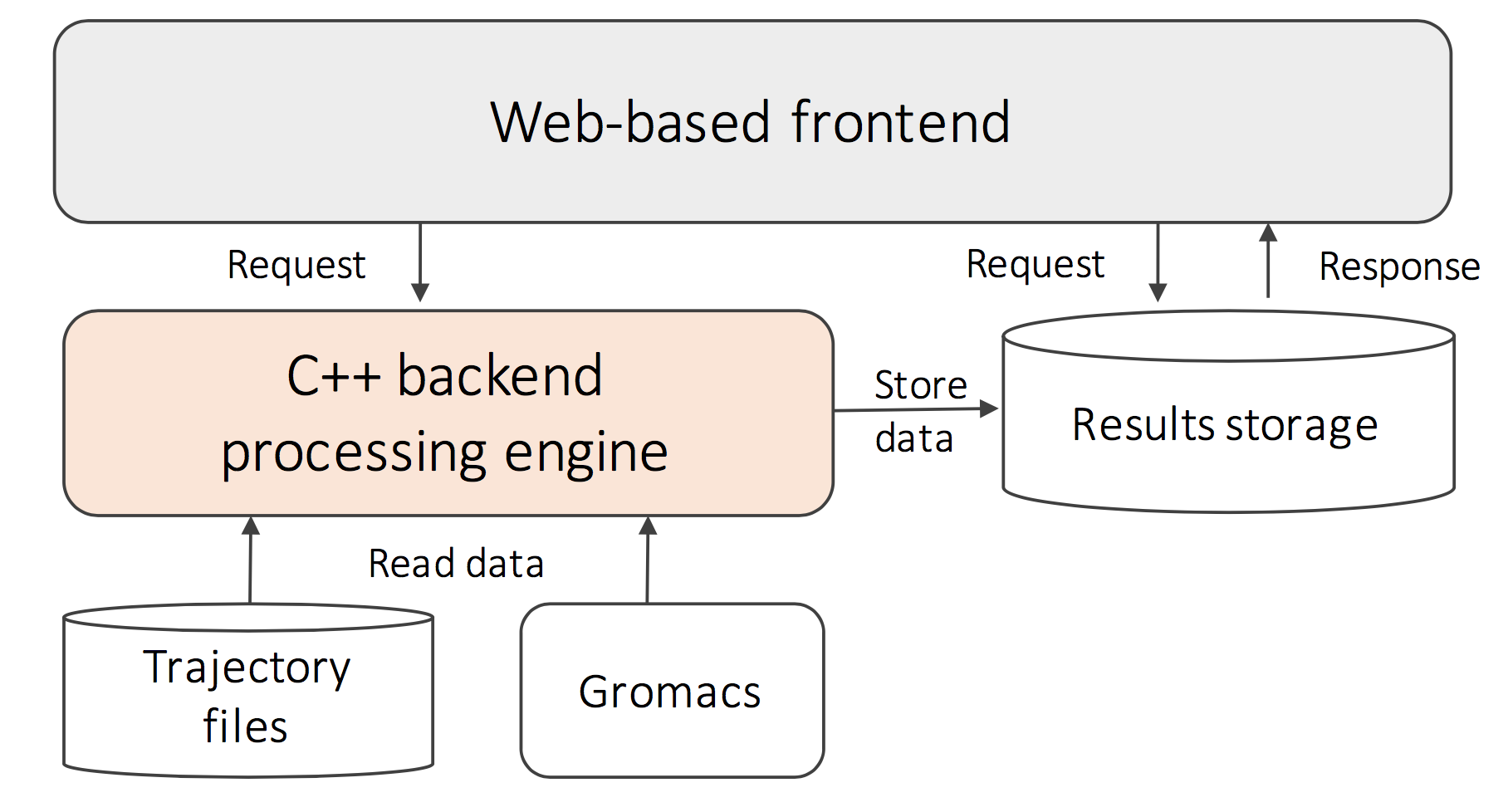
MSanalysis comes with an easy-to-use GUI as the fron- tend, and a powerful C++ based processing engine as backend (see the following figure). The user interface is a web application written by python. Due to its cross- platform compatibility, users can access MSanalysis through a browser on different operating systems. The processing engine is a standalone program written by C++ , which is closer to hardware and designed for efficient execution. The processing engine is designed following a push-based data processing model, which is very efficient to handle the analysis with multiple queries.
MSanalysis accepts two flavors of data inputs: (1) trajectory files generated from a molecular simulation program. Currently, MSanalysis supports files following the GROMACS format, and more such formats will be supported in the future. (2) MSanalysis can also read in the simulation results from GROMACS on-the-fly as the simulation is underway through a data transmission channel. In this mode, a small software module for data transmission needs to be installed in the GROMACS program.
The analysis results of MSanalysis include two parts. The first part is the information used to identify an analysis, including the data source, queries included, and the selection information. This information will be stored in a database, which facilitates history records checking. The second part is the results of the processed queries, which are stored in different files related to different analysis. We also provide basic visualization of this part of the output via the GUI.
© 2016-2025 MSanalysis